Behçet disease, or Behçet syndrome, is a chronic, relapsing, inflammatory disorder of unknown etiology. It is a multisystem disease that is characterized by oropharyngeal and genital ulcers and ocular involvement, and there may be vascular, articular, gastrointestinal, neurologic, urogenital, pulmonary, and cardiac findings as well. In genetically predisposed individuals, it may be triggered by infectious agents such as herpes simplex 1 (HSV-1) or Streptococcus; histamine-releasing foods such as citrus fruits, cheese, or nuts; poor oral hygiene; or stress; or triggers may not be identified. Circulating immune complexes and neutrophils are part of the pathogenesis, leading to vascular injury.
Behçet disease is relatively uncommon and is more prevalent in Japan and Middle Eastern and Mediterranean countries. While Behçet disease has an equal sex distribution overall, there is male predominance in Middle Eastern and Mediterranean patients and female predominance in Japan and Korea. Severe disease is more commonly seen in males. The onset of symptoms typically occurs during the third or fourth decades of life. Rarely, it can present during childhood, and usually in children with a positive family history of the syndrome. Behçet disease is strongly associated with the HLA-B51 allele, which is present in more than 80% of Asian patients with Behçet disease.
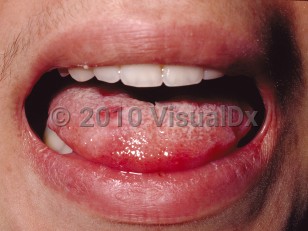
Clinically, Behçet disease is characterized by recurrent oropharyngeal and genital ulcers and ocular involvement. Oral ulcers are the initial presenting sign in up to 80% of patients and can lead to scarring, dysphagia, and odynophagia. Genital ulcers present similarly to oropharyngeal lesions; they tend to recur less frequently but are more prone to scar formation. Potential complications include epididymo-orchitis.
Ocular disease, seen more often in men, is characterized by episodes of bilateral, nongranulomatous anterior and/or posterior uveitis. It usually appears 2-3 years after the onset of oral and/or genital ulcers, although it can be the initial manifestation in up to 20% of cases. Approximately 1 in 4 patients with ocular disease develop blindness.
Other cutaneous manifestations include erythema nodosum-like lesions, folliculitis-like lesions, erythema multiforme-like lesions, Sweet syndrome-like lesions, subcutaneous thrombophlebitis, and palpable purpura.
Behçet disease can have significant morbidity and mortality, with major causes including neurologic, vascular, and gastrointestinal involvement. Patients may experience remission, however, after years of active disease.
Related to Behçet syndrome is Hughes-Stovin syndrome, in which patients don't have the clinical criteria for Behçet syndrome but have a vasculitis and may have pulmonary aneurysm.
Behçet syndrome - Anogenital in
See also in: Overview,Oral Mucosal LesionAlerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
M35.2 – Behçet's disease
SNOMEDCT:
310701003 – Behcet's syndrome
M35.2 – Behçet's disease
SNOMEDCT:
310701003 – Behcet's syndrome
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:04/15/2019
Last Updated:03/06/2024
Last Updated:03/06/2024
Behçet syndrome - Anogenital in
See also in: Overview,Oral Mucosal Lesion