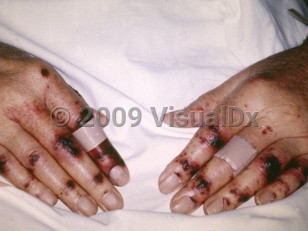
Thrombotic thrombocytopenic purpura (TTP) can be a severe and life-threatening disease characterized by microangiopathic hemolytic anemia, thrombocytopenia, and organ dysfunction.
TTP is a subtype of the thrombotic microangiopathy (TMA) syndromes. Other subtypes of TMA include Shiga toxin-mediated TMA (Shiga toxin hemolytic-uremic syndrome), drug-mediated TMA, and complement-mediated TMA, among others. Accurate diagnosis of TTP and prompt initiation of treatment are essential to prevent morbidity and mortality.
The cause may be acquired or, in rare cases, hereditary (Upshaw-Schulman syndrome). In acquired TTP, also known as immune-mediated TTP (iTTP), autoantibodies form against the plasma metalloprotease ADAMTS13, which normally cleaves von Willebrand factor (vWF). As a result, vWF multimers accumulate, causing platelet aggregation and microvascular occlusion. Hereditary TTP results from genetic mutations in the ADAMTS13 gene, resulting in the absence or severe deficiency of ADAMTS13.
Classically, TTP was thought to be associated with a "pentad" of clinical and laboratory findings including microangiopathic hemolytic anemia, thrombocytopenia, neurologic abnormalities, fever, and renal dysfunction. However, the latter 3 findings are not reliably present in TTP, and consequently the presence of microangiopathic hemolytic anemia and thrombocytopenia are sufficient for a diagnosis of TTP.
Clinical features of TTP are varied and may include gastrointestinal symptoms (abdominal pain, nausea, vomiting), fatigue, malaise, neurologic abnormalities (altered mental status, confusion, headache), and purpuric rash.
Due to its rarity and variety of ADAMTS13 mutations, there is still much to learn about hereditary TTP. Patients with hereditary TTP can present at birth or remain asymptomatic into adulthood. Specific signs and symptoms may be related to the severity of ADAMTS13 mutations, with more severe cases related to lower ADAMTS13 activity. When symptoms first manifest, presentation can be quite variable and may be similar to an acquired TTP episode.
Although patients with hereditary TTP may remain asymptomatic for many years, their increased risk of critical thrombosis is always present. Overt thrombotic episodes in patients with hereditary TTP are associated with states of increased vWF concentrations, such as during pregnancy or the neonatal period or due to trauma, excessive alcohol intake, infection, or inflammation. However, clinically silent infarcts may occur outside of such states.
TTP is more common in women and in Americans of African descent. Hereditary TTP is usually initially recognized before age 10 or during a woman's first pregnancy. Acquired TTP typically presents between the ages of 30 and 50. For acquired TTP, overall survival rates with therapy are above 80%, although relapse occurs in up to 40% of cases.
Potentially life-threatening emergency
Thrombotic thrombocytopenic purpura
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
M31.19 – Other thrombotic microangiopathy
SNOMEDCT:
78129009 – Thrombotic thrombocytopenic purpura
M31.19 – Other thrombotic microangiopathy
SNOMEDCT:
78129009 – Thrombotic thrombocytopenic purpura
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
Drug Reaction Data
Subscription Required
References
Subscription Required
Last Reviewed:04/29/2020
Last Updated:03/12/2024
Last Updated:03/12/2024
Potentially life-threatening emergency
Thrombotic thrombocytopenic purpura