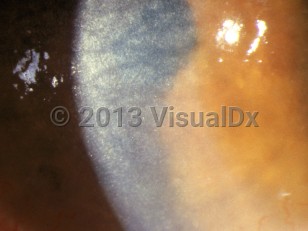
Cystinosis is a rare, autosomal recessive lysosomal storage disease caused by a defective transporter protein for cystinosin that leads to a pathologic accumulation of intracellular lysosomal cystine, resulting in crystalline formation that damages organs and tissues throughout the body. While all cells and organs are affected, the kidneys, eyes, thyroid, pancreas, testes, and heart are most vulnerable to damage. There are 3 main forms of cystinosis: infantile (early-onset or nephropathic), intermediate (late or juvenile / adolescent onset), and ocular (non-nephropathic).
Infantile (early-onset) cystinosis is the most severe form of cystinosis, resulting in failure to thrive and renal Fanconi syndrome with symptoms beginning in infancy. Symptoms include polyuria, polydipsia, acidosis, dehydration, hypophosphatemic rickets, and electrolyte imbalances. If untreated, renal failure may ensue by the age of 10 years. The thyroid gland is often affected, resulting in hypothyroidism. Pancreatic endocrine failure results in the development of diabetes mellitus, with testicular involvement leading to possible infertility and hypogonadism. Ocular involvement often results in visual acuity deterioration that can lead to blindness as well as photophobia. Muscles may also become involved, resulting in weakness and wasting. This may impair the ability to ambulate as well as affect functions such as swallowing. Brain involvement results in impaired attention, focus issues, and memory issues, as well as incoordination. Gastrointestinal (GI) involvement can lead to hepatosplenomegaly (HSM), poor appetite, and recurrent vomiting.
The intermediate form of cystinosis does not become symptomatic until adolescence, with involvement of mainly the kidneys and eyes. If not appropriately managed, renal failure will often occur by the patient's late teens or mid-20s. While other organ system involvement occurs, significant dysfunction is seen much less frequent than in early-onset disease.
The diagnosis of cystinosis is suspected based on symptoms and/or family genetic history. Ocular crystalline deposits are not usually seen until after 1 year of age. Genetic testing and WBC cystine content measurements are the best tests to confirm the diagnosis.
There is no current cure for cystinosis. Treatment consists of using cystine-depleting medications and careful monitoring of cystine concentrations in WBC as well as treatment for organ dysfunction (eg, dialysis / renal transplantation, diabetes management, treatment of hypothyroidism).
Cystinosis
See also in: External and Internal EyeAlerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
E72.04 – Cystinosis
SNOMEDCT:
190681003 – Cystinosis
E72.04 – Cystinosis
SNOMEDCT:
190681003 – Cystinosis
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:01/30/2024
Last Updated:02/06/2024
Last Updated:02/06/2024