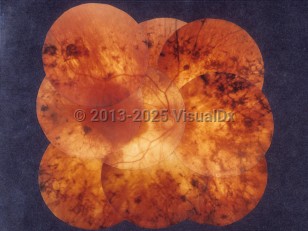
Retinitis pigmentosa (RP) is a heterogeneous group of inherited retinal disorders that predominantly affects the retinal photoreceptors, resulting in a progressive visual decline. It is characterized as a progressive, diffuse vision loss that affects mainly night vision and peripheral vision. It starts with rod photoreceptor degeneration followed by cone photoreceptors and retinal pigment epithelial (RPE) cell degeneration. Prevalence of RP is 1 in 3000-5000 people. Because the rods are affected first, patients will start to have trouble with night vision. As the disease progresses over decades, the peripheral vision loss can progress to tunnel vision. The disease is typically bilateral but can be asymmetric.
The clinical picture may occur in isolation or as part of numerous syndromic conditions. These include abetalipoproteinemia, Usher syndrome, Biemond syndrome type 2, Jalili syndrome, Senior-Løken syndrome, Bardet-Biedl syndromes, disorders of glycosylation, Kearns-Sayre syndrome, gyrate atrophy, and neurodegeneration with brain iron accumulation (formerly Hallervorden-Spatz disease).
There is no known risk factor to RP. Many gene mutations are associated with it, the most common being the rhodopsin gene (RHO). RP can occur as a sporadic mutation or be autosomal dominant, recessive, or X-linked. Most autosomal recessive cases are associated with other systemic disorders. Patients can undergo genetic testing to see which gene mutations are associated with which disease. Mutation in RPE65 is particularly important as that mutation is the only one approved for genetic therapy.
True RP is a nonsyndromic ocular disease with a characteristic clinical picture as described here, while many of the pigmentary changes in the fundi of individuals with associated syndromes are more variable and should be called "pigmentary retinopathy" instead.
Retinitis pigmentosa - External and Internal Eye
Synopsis

Codes
ICD10CM:
H35.52 – Pigmentary retinal dystrophy
SNOMEDCT:
28835009 – Retinitis pigmentosa
H35.52 – Pigmentary retinal dystrophy
SNOMEDCT:
28835009 – Retinitis pigmentosa
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:08/12/2020
Last Updated:01/23/2022
Last Updated:01/23/2022