Junctional epidermolysis bullosa (JEB) is 1 of the 4 major types of inherited epidermolysis bullosa (EB). The other major types are EB simplex, dystrophic EB, and Kindler syndrome. JEB is a genodermatosis characterized by inherently fragile skin that blisters with trauma. JEB results from autosomal recessive mutations within one of the genes encoding structural proteins within the lamina lucida of the skin basement membrane zone (BMZ) or within the hemidesmosome. These proteins are responsible for cell adhesion.
All patients with the most severe subtype of generalized JEB, severe JEB (previously known as JEB generalized severe, Herlitz JEB), have mutations within any of the 3 genes (LAMA3, LAMB3, or LAMC2) corresponding to the 3 subunits of the extracellular matrix protein laminin-332 (previously laminin-5). Similarly, the majority of JEB patients with generalized but clinically less severe disease, intermediate JEB (previously named JEB generalized intermediate, non-Herlitz JEB), have structurally less severe mutations within the same 3 laminin-332 genes. A minority of intermediate cases are caused by mutations in the gene that encodes type XVII collagen. Rare subtypes of JEB may have mutations in genes encoding other structural proteins: JEB with pyloric atresia (JEB-PA) is associated with mutations in either of the genes encoding the 2 subunits of a6b4 integrin. A rare genodermatosis named the laryngo-onycho-cutaneous syndrome (LOC syndrome, previously named Shabbir syndrome), which was recently included among the subtypes of JEB, is notable for having mutations within the gene encoding for the a3 chain of laminin-332. Autosomal recessive mutations of integrin alpha-3 (ITGA3) are associated with junctional skin lesions, interstitial skin disease, and nephrotic syndrome.
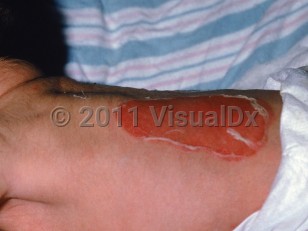
The 3 most common subtypes of JEB are severe JEB, intermediate JEB, and JEB inversa, the latter of which is characterized by cutaneous disease primarily localized symmetrically in body folds (ie, axillary and inguinal vaults and perineum), the nape of the neck, and the lumbosacral area. Both severe and intermediate JEB are usually associated with generalized blisters, erosions, atrophic scars, and extensive postinflammatory hypo- or depigmentation. A characteristic finding in severe JEB is the development of thick, exuberant, periorificial granulation tissue that may also appear within the nares, upper back, nape of the neck, and periungual folds. Recurrent granulation tissue may totally occlude the nasal passage or progress into the upper airway.
Patients with severe JEB have profound growth retardation and severe multifactorial anemia. The oral cavity and upper esophagus are often severely affected in severe JEB as well as in JEB inversa. Findings may include marked microstomia, ankyloglossia, and primary enamel hypoplasia, the latter resulting in pitting of the teeth, severe secondary caries, and eventual premature loss of teeth. Esophageal stenoses or strictures may occur in JEB, similar to those seen in severe generalized recessive dystrophic EB. Scarring alopecia of the scalp and delayed puberty are common features of severe JEB. About 3% of these patients develop partial webbing deformities of the hands or feet compared with patients with severe generalized recessive dystrophic EB, who invariably get this deforming musculoskeletal complication.
Intermediate JEB can be contrasted to severe JEB by the infrequency of clinically significant growth impairment or anemia, lack of exuberant granulation tissue, and relatively mild intraoral disease activity.
Since the characteristic distinguishing clinical features of severe and intermediate JEB may not become obvious for the first 1-2 years of life, some authorities prefer to classify those children as having a "JEB indeterminant subtype" pending more definitive clinical or laboratory data allowing more precise subclassification and prognostication.
Approximately 40% of individuals with severe JEB do not survive to adolescence. Mortality from severe and intermediate JEB is high during the first few years of life, predominantly from sepsis, failure to thrive, and/or upper airway obstruction. Approximately half of all children with JEB enrolled in the National (United States) EB Registry died within their first year of life. Tracheolaryngeal obstruction is a major threat in both of these JEB subtypes; the risk may persist until age 6 years. The earliest sign of tracheolaryngeal involvement is a persistent hoarse or weak cry. If progressive, stridor and sudden (and often fatal) airway obstruction may occur. Extreme vigilance and early intervention for this complication is essential.
As opposed to recessive dystrophic EB, development of squamous cell carcinomas or renal failure (see acute renal failure and chronic renal failure) are less commonly seen in JEB, likely due to death in early childhood, although there are reports of cutaneous squamous cell carcinomas in adults with intermediate type.
The eyes may also be involved in both severe and intermediate JEB, resulting in acute onset of severe pain and blistering. If untreated, corneal scarring and even blindness may rarely ensue.
JEB-PA (associated with pyloric atresia) is a very rare EB subtype that can be readily diagnosed during the first day of life on the basis of clinical and radiological findings of pyloric atresia. Some of these patients also have a variety of genitourinary tract anomalies.
The cutaneous involvement in LOC syndrome (laryngo-onycho-cutaneous syndrome) predominantly involves the facial and neck regions and is associated with severe involvement of the larynx and conjunctiva.
Revertant mosaicism, when a second somatic or postzygotic mutation results in a functional protein, is most commonly reported in intermediate JEB but has been reported in all EB subtypes.
Related topics: generalized severe EB simplex, EB acquisita
Junctional epidermolysis bullosa in Child
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
Q81.8 – Other epidermolysis bullosa
SNOMEDCT:
399971009 – Junctional epidermolysis bullosa
Q81.8 – Other epidermolysis bullosa
SNOMEDCT:
399971009 – Junctional epidermolysis bullosa
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:03/27/2024
Last Updated:04/08/2024
Last Updated:04/08/2024
Junctional epidermolysis bullosa in Child