Phenylketonuria (PKU) is an autosomal recessive disorder of phenylalanine metabolism. PKU screening is now part of standard neonatal screening in most countries for inborn errors of metabolism. Accumulation of phenylalanine and its metabolites are thought to be responsible for the pathologies seen in PKU.
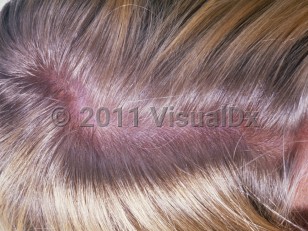
The untreated state is characterized by a constellation of functional and physical manifestations including intellectual disability, a characteristic bodily appearance, and neurological impairments. Intellectual disability worsens from early childhood to adolescence during brain maturation. Patients develop the classic triad of blond hair, blue eyes, and light skin usually by the second year of life. Atopic dermatitis may occur, and sclerodermatous changes limited to skin and connective tissue have been reported. Patients may also have neuropsychiatric abnormalities.
Phenylketonuria in Child
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
E70.0 – Classical phenylketonuria
SNOMEDCT:
7573000 – Classical phenylketonuria
E70.0 – Classical phenylketonuria
SNOMEDCT:
7573000 – Classical phenylketonuria
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Updated:01/20/2022